Enrichment Analysis
Overview
Enrichment analysis aim to find functional set which are over-represented in the large set of protein or genes. In this section, we will go through the supported enrichment methods, demonstrate the definition and the usage of each method.
Basic class
All the class of enrichment analysis are inherited from EnrichmentResult. This class holds the
result of the enrichment analysis and the subclass should at least
rewrite the statistic method run to perform the analysis.
methods
-
run: execute the enrichment analysis and return the instance of subclass contains the analysis information -
table: return the resultpandas.Dataframe -
plot: plot the bar chart in the output area -
graph: if the result set have certain relationship, this method return the graph of them. for example, the Gene Ontology ORA subclass return the graph of GO DAG of significant sets.
ORA
The over-representation analysis is the most widely used enrichment analysis method, It use Fisher's exact test to check whether certain functional set is over-represented in a large set of protein or genes.
Implementation
The ORA methods is implemented in the analysis.ora
# general ora method
ORA.run(study, pop, gene_set, adjust='fdr_bh')
Parameters
study:setorlistof significant differential expressed genes or proteins. E.g.[A, B, C, D]popsetorlistof the total annotated genes or proteins. E.g.[A, B, C, D, E, F, G]gene_setis a dict contains. the gene set name and contained genes like
{"B cell receptor pathway": {"CD22", "CD81"...}...}
This information usually parsed from a GMT file use GMTUtils.
Example
# import necessary module
from pypathway.analysis.ora import KEGG, ORA
from pypathway.utils import ColorectalCancer, IdMapping, GMTUtils
# load a gene_set from a gmt file
# the test folder does not come with pypi or anaconda installation, please obtain it from GitHub.
# git clone https://github.com/iseekwonderful/PyPathway
gmt = GMTUtils.parse_gmt_file("../../tests/assets/gmt_file/h.all.v6.0.entrez.gmt")
# load data set
c = ColorectalCancer()
# perform general ORA test
res_h = ORA.run(c.deg_list, c.background, gmt)
# view result table
res_h.table.head()
The result table (In IPython notebook):
| name | mapped | number in study | p-value | fdr | |
|---|---|---|---|---|---|
| 0 | HALLMARK_GLYCOLYSIS | 184 | 91 | 9.956407e-08 | 3.555860e-07 |
| 1 | HALLMARK_APICAL_JUNCTION | 185 | 97 | 7.750142e-10 | 3.748583e-09 |
| 2 | HALLMARK_MYC_TARGETS_V1 | 173 | 48 | 8.377692e-01 | 9.106187e-01 |
| 3 | HALLMARK_COAGULATION | 131 | 64 | 1.237516e-05 | 3.093790e-05 |
| 4 | HALLMARK_MTORC1_SIGNALING | 176 | 90 | 1.551015e-08 | 5.965443e-08 |
# barplot
res_h.plot()

Specific ORA
For KEGG, REACTOME and GO, there are specific class implement for them. This class preloaded the gene sets or the DEG file before the analysis are performed.
The APIs
The input and output data structure are same as the basic ORA.
KEGG
#rather than input specific gene set, organism is accepted and geneset will be retrieved.
r_kg = KEGG.run(c.deg_list, c.background, 'hsa')
Reactome
# Reactome official API is used. note that the background is the whole Reactome
# library, if background set required, use Reactome GMT file and ORA class
r = Reactome.run(sybs, organism='Homo sapiens')
Gene Ontology
For Gene Ontology analysis please provide a association file, which could be a Python dict or text file. In this example, we use a dict generated by the utility IdMapping. If you want use the exist text association file, use the format like this each line: ID\tGO_term1;GO_term2...\n
# Gene ontology analysis require additional a assoc file.
# IdMapping class could be used to generate this file
# c.background is to background gene list of certain study
r = IdMapping.convert_to_dict(input_id=c.background, source='ENTREZID', target="GO", organism='hsa')
# and run the analysis
# [str(x) for x in c.deg_list]: study
# [str(x) for x in c.background]: background
# r: the assoc dict
# obo=path + 'go-basic.obo': the path to the obo file
rg = GO.run([str(x) for x in c.deg_list], [str(x) for x in c.background], r, obo=path + 'go-basic.obo')
The gene ontology enrichment analysis provide the graph overview of the analysis

GSEA
The Gene Set Enrichment Analysis(GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states. is introduced in paper:
Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles
at PNAS
This class is implement based on GSEApy
Usage
Input data structure
Differ to other analysis class, the GSEA need to do a phenotypic or gene_set permutation test, so the original data is need.
-
The gene_sets: receives both get dict or a library name in Enrichr's library
-
The cls file, this file format defines phenotype (class or template) labels and associates each sample in the expression data with a label. The CLS file format uses spaces or tabs to separate the fields. For detail information, please refer to the official documents
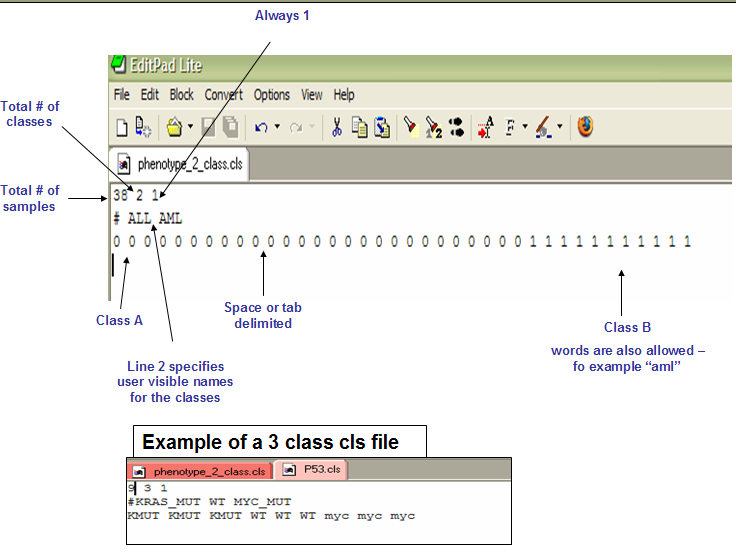
- The expression file. A pandas data frame contains the expression value (counts, TPM). e.g.
| Name | 2965M patient's mucosa | 3216M patient's mucosa | 3335M patientüs mucosa | 3416M patient's mucosa | 3578M patientüs mucosa | 3798M patientüs mucosa | 3838M patientüs mucosa |
|---|---|---|---|---|---|---|---|
| A1BG-AS1 | 1.618062 | 1.631023 | 1.751713 | 1.942189 | 1.389749 | 1.620180 | 1.222842 |
| A1CF | 2.029520 | 3.323313 | 3.315563 | 2.795794 | 3.209412 | 2.805799 | 1.633054 |
| A2M | 2.994217 | 2.556958 | 2.999796 | 3.354563 | 3.143560 | 2.776570 | 3.075487 |
| A2M-AS1 | 2.233742 | 1.709143 | 1.968936 | 2.648101 | 2.353158 | 2.346996 | 2.903206 |
API
# gene_exp is the expression dataframe
# class_vector is the define of the experiment
r = GSEA.run(data=gene_exp, gmt="KEGG_2016", cls=class_vector)
more detail information is available in the notebook
SPIA
The Signaling Pathway Impact Analysis, is introduced in paper:
We implement this algorithm in Python.
Usage
r = SPIA.run(de=c.deg, all=c.background)
Parameters
- de: a python dict of DEGs.
key: gene,value: fold-change. e.g.{'A': 2.1, 'B': 3.0 ...} - all: a python list of total genes. the idtype is ENTREZ. If necessary, use IDMapping to convert other idtype to ENTREZ. e.g.
['A', 'B', 'C', 'D']
Result
Use table method to view the result in table form and use the plot method to view the bar plot.
Example notebook is also available.
Enrichnet
The Enrichnet algorithm is introduced in paper
We implement the Python API for Enrichnet HTTP service.
Usage
run(genesets, idtype='hgnc_symbol', pathdb='kegg', graph='string')
Parameters
- genesets: the gene list used to run the analysis. E.g.
['A', 'B' ...] - idtype: input id type. Supports idtype:
['ensembl', 'hgnc_symbol', 'refseq_dna', 'uniprot_swissprot'] - the pathlib: function set library. Supports pathdb:
['kegg', 'biocarta', 'reactome', 'wiki', 'nci', 'interpro', 'gobp', 'gomf', 'gocc'] - graph: the graph database. Supports
['string', 'bossi'].
Result
Use table method to view the result in table form and use the plot method to view the bar plot.
Example notebook is also available.